Transposition der großen Arterien (TGA)
Die Transposition der großen Arterien stellt den zweihäufigsten angeborenen Herzfehler mit Blausucht dar. Bei diesem angeborenen Herzfehler sind die beiden großen Schlagadern des Herzens – die Aorta und die Pulmonalarterie – vertauscht (also „transponiert“). Es handelt sich um eine lebensbedrohliche Fehlbildung, die unmittelbar nach der Geburt behandelt werden muss.
Definition
Die Transposition der großen Arterien (TGA) bezeichnet die inkorrekte Lage der beiden großen Gefäße, d.h. die Körperschlagader (Aorta) und die Lungenschlagader (Pulmonalarterie) stehen falsch zueinander und zu den Pumpkammern des Herzens, von denen diese entspringen. Es gibt verschiedene Varianten dieser Transposition: eine totale Transposition, eine partielle Transposition oder eine korrigierte Transposition. Patient:innen können zusätzlich an einem weiteren Herzfehler erkranken.
Wir beschreiben im Folgenden die totale Transposition. Bei dieser Form der Erkrankung ist der Ursprung der beiden großen Gefäße vertauscht. Die Körperschlagader entspringt an der Stelle, an der sich beim gesunden Herzen die Lungenschlagader befindet, also von der rechten Herzkammer ausgehend. Umgekehrt kommt die Lungenschlagader aus dem linken Herzen.
Die übrigen Strukturen des Herzens sind normal ausgebildet und haben keine weiteren Anomalien, deshalb nennt man sie auch „einfache Transposition“, wobei die Patient:innen auch in diesem Fall sofort nach der Geburt in einer kinderherzmedizinischen Spezialklinik versorgt werden müssen. Die Kinder werden zwar termingerecht geboren und haben ein normales Aussehen. Ihre Haut verfärbt sich jedoch schon in den ersten Lebensstunden bläulich.
Ursachen
Bisher konnten keine typische strukturelle Veränderung des Chromosoms für die d-TGA identifiziert werden. Es werden Spontanmutationen für die Erkrankung verantwortlich gemacht. Das genetische Risiko bei Kindern von Eltern mit einer Transposition der großen Arterien oder bei einem betroffenen Geschwisterkind ist gering und kann nicht in Prozent angegeben werden, da nur wenige Einzelfallberichte vorliegen.
Zusätzlich wird über eine Häufung bei mütterlichem Diabetes mellitus, Alkoholkonsum während der Schwangerschaft und Mangelernährung berichtet.
Folgen
In einem normalen Herzen fließt sauerstoffreiches Blut aus den Lungen in den linken Vorhof und die Kammer und wird von dort durch die Aorta in den ganzen Körper gepumpt, der dadurch mit Sauerstoff versorgt wird. So entwickelt sich sauerstoffarmes Blut, welches dann zurück in den rechten Vorhof und die rechte Kammer gelangt und von dort über die Lungenschlagader in die Lunge gepumpt wird, um dort wieder mit Sauerstoff angereichert zu werden. Das bedeutet, dass der Lungenkreislauf und der Körperkreislauf in Serie, also „nacheinander“ geschaltet und damit verbunden sind.
Bei der Transposition gelangt das venöse Blut jedoch in den rechten Vorhof und die rechte Kammer, anschließend jedoch wieder über die Aorta in den großen Kreislauf, ohne durch die Lungen zu fließen und mit Sauerstoff angereichert zu werden. Dies führt zu Sauerstoffarmut im Blut. Das Blut aus den Lungen kommt stattdessen über den linken Vorhof und die linke Kammer wieder zurück über die Lungenschlagader in die Lunge. Dadurch entstehen zwei getrennte Kreisläufe, die nicht miteinander verbunden sind – das Kind bekommt in der Folge nicht genug Sauerstoff.
Einzig ein geringer Fluss in das linke Herz und von dort in die Lungen ist noch möglich, weil die kleine natürliche Verbindung zwischen den beiden Vorhöfen offenbleibt. Dies ist die einzige Verbindung zwischen den beiden Kreisläufen. Die Sauerstoffmenge, die durch dieses kleine Loch transportiert wird, ist jedoch in den meisten Fällen nicht ausreichend zum Weiterleben. Daher ist die totale Transposition für diese Patient:innen lebensbedrohlich. Die Kinder befinden sich bereits kurz nach der Geburt in einem kritischen Zustand und benötigen dringend einen kinderherzchirurgischen Eingriff.
Symptome
Bei Kindern mit totaler Transposition der großen Arterien kommt es schon in den ersten Lebensstunden zur Blaufärbung der Haut (Zyanose). Patient:innen leiden außerdem an schneller Atmung, Lethargie und einer lebensbedrohlichen Sauerstoffunterversorgung.
Diagnose
Die Transposition der großen Arterien (TGA) lässt sich am besten per Echokardiographie (Herzultraschall) feststellen. Oft ist die Erkrankung schon während der Schwangerschaft erkennbar.
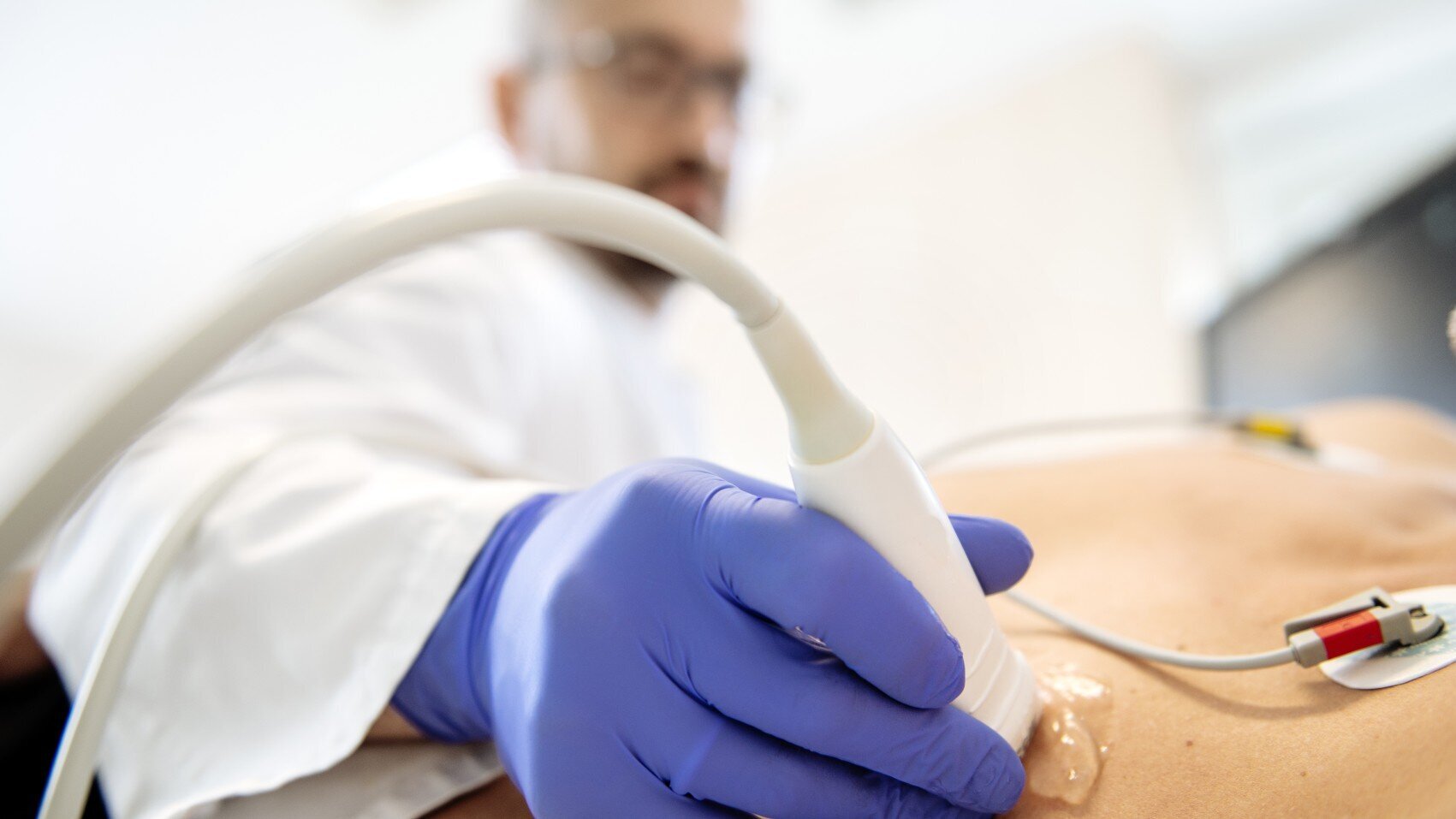
Die Transposition der großen Arterien (TGA) lässt sich am besten per Echokardiographie (Herzultraschall) feststellen. Oft ist die Erkrankung schon während der Schwangerschaft erkennbar.
Therapie
In einem ersten Schritt besteht die Möglichkeit, das kleine Loch zwischen den Vorhöfen zu vergrößern, um auch den Fluss in die Lungen zu erhöhen. Dies gelingt meist mit einem Herzkathetereingriff (Rashkind-Manöver). Das bedeutet, dass durch das zu kleine Loch ein Katheter mit Ballon geschoben und dann aufgelassen wird, wodurch sich das Loch vergrößert. Dieser Eingriff muss meist als dringlicher Notfall durchgeführt werden. Diese Vergrößerung des Lochs und der dadurch erhöhte Fluss in die Lungen verbessern den Zustand des Kindes und schaffen Zeit, um die Operation zu planen.
Der Goldstandard der Therapie der Transposition der großen Gefäße ist die anatomische Korrektur. Das bedeutet die chirurgische Umplatzierung und Verbindung der Aorta mit der „eigenen“ linken Pumpkammer und der Lungenschlagader mit der „eigenen“ rechten. Dabei werden die Aorta und Pulmonalarterie an ihre richtigen Plätze versetzt. Meist geschieht dieser Eingriff innerhalb der ersten ein bis zwei Lebenswochen.
Nach dem Öffnen des Brustkorbes wird die Patientin bzw. der Patient an die Herz-Lungen-Maschine angeschlossen, die für die Zeit des Eingriffs die Herz- und Lungenfunktion übernimmt. Gleichzeitig wird der Körper von außen und parallel das Blut an der Herz-Lungen-Maschine gekühlt. Bei Kühlung wird der Sauerstoffverbrauch bei allen Organen deutlich reduziert – das bietet Schutz gegen mögliche postoperative Komplikationen.
In der Operation werden die vertauschten großen Arterien durchtrennt und in der korrekten Platzierung wieder angenäht. Dazu müssen die Gefäße freigelegt und der PDA durchtrennt werden. Entscheidend ist, dass die Ursprünge der Herzkranzgefäße zusammen mit der Aorta versetzt werden, damit der Herzmuskel auch mit sauerstoffreichem Blut versorgt wird. Diese kleinen Gefäße werden aus der Wand herausgetrennt und dann in die verpflanzte Aorta wieder eingenäht. Die Entnahmestellen der Herzkranzgefäße werden mit Flicken wieder ausgefüllt. Danach wird die Lungenarterie vor die Aorta gebracht, sodass die Gefäße wieder aneinandergenäht werden können. Diese Operation heißt „arterielle Switch-Operation“.
Viele Operationen am Herzen können nur am stillstehenden Herzen durchgeführt werden. Eine Herz-Lungen-Maschine übernimmt während der OP die Funktion von Herz und Lunge.
Am DHZC stehen zwölf moderne Herz-Lungen-Maschinen zur Verfügung, drei davon speziell für Säuglinge und Kleinkinder.
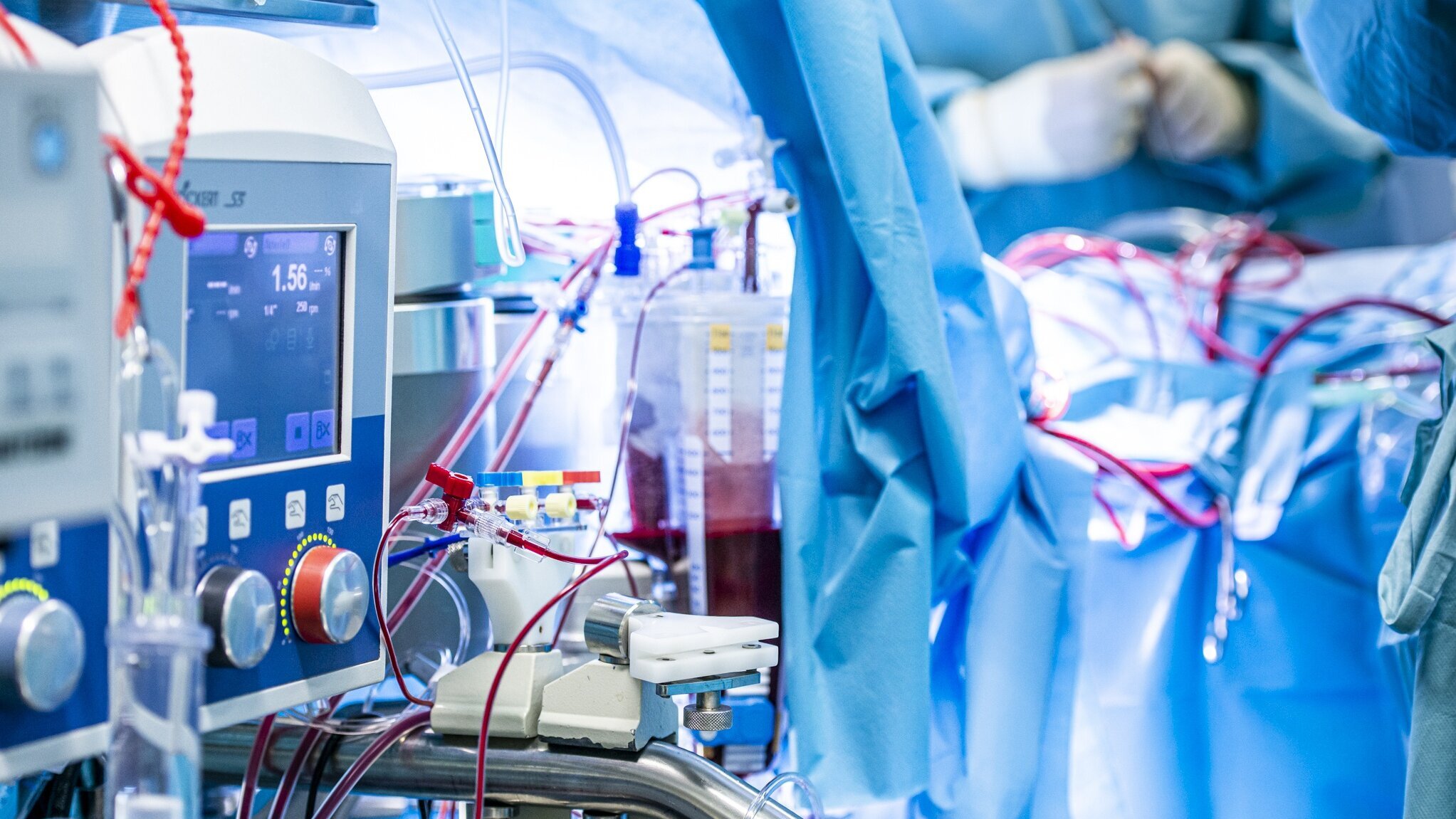
Viele Operationen am Herzen können nur am stillstehenden Herzen durchgeführt werden. Eine Herz-Lungen-Maschine übernimmt während der OP die Funktion von Herz und Lunge.
Am DHZC stehen zwölf moderne Herz-Lungen-Maschinen zur Verfügung, drei davon speziell für Säuglinge und Kleinkinder.
Therapie am DHZC
Die Herz-Lungen-Maschine mit dem weltweit kleinstem Füllvolumen gibt uns viel Chancen, diese komplizierte Operation an Neugeborenen ohne Fremdblut-Konserven durchführen zu können, was für die Babys erhebliche Vorteile hat. Damit können wir nicht nur die Infektions- und Unverträglichkeitsrisiken minimieren, sondern unseren kleinen Patient:innen häufig auch eine schnellere Erholung nach der Operation ermöglichen.
Zur Kinderherzchirurgie am DHZC
Diagnostische Herzkatheteruntersuchungen am DHZC
In zwei speziell ausgerüsteten und hochmodernen Herzkatheterlaboren der Klinik für Angeborene Herzfehler – Kinderkardiologie untersuchen und behandeln wir Neugeborene, Säuglinge, Kinder und Jugendliche jeden Alters mit angeborenen und erworbenen Herzerkrankungen, sowie auch alle Erwachsenen mit angeborenen Herzfehlern. Wir sind als überregionales Schwerpunktzentrum auf die invasive kardiologische Diagnostik und interventionelle Therapie von Patient:innen mit angeborenen Herzfehlern spezialisiert. Interventionelle Behandlungen im Herzkatheterlabor ergänzen oder ersetzen zunehmend chirurgische Eingriffe und sind entscheidend, um die optimale Therapie der Patient:innen festzulegen. Unser Team verfügt über langjährige Erfahrung und Expertise.
Fragen und Antworten für Eltern (FAQ)
Nach der Operation kann sich Ihr Kind ohne Risiko normal belasten. In einzelnen Fällen können Medikamente bei der Entlassung notwendig sein.
Regelmäßige und lebenslange Kontrolluntersuchungen durch eine:n auf angeborene Herzfehler spezialisierte:n Ärztin/Arzt sind erforderlich.
Langzeitergebnisse zeigen, dass sich die Kinder in der Regel normal entwickeln.
Mehrere Arbeiten zum Langzeitverlauf mit großer Patientenzahl zeigen, dass die Herzkranzgefäße mitwachsen und nur fünf bis neun Prozent der Patient:innen erneut operiert werden müssen.
Sehr selten (in zwei bis fünf Prozent der Fälle) treten Aortenklappeninsuffizienzen auf, die zu einem Klappenersatz führen.
Autorinnen

Dr. med. Antonia Schulz ist Oberärztin der Klinik für Chirurgie Angeborener Herzfehler – Kinderherzchirurgie am Deutschen Herzzentrum der Charité (DHZC) mit Schwerpunkt auf minimalinvasiver Chirurgie. Seit 2022 ist sie Fellow des BIH Charité Digital Clinician Scientist Programms.

Dr. med. Kira Kuschnerus arbeitet als Fachärztin an der Klinik für Chirurgie Angeborener Herzfehler – Kinderherzchirurgie am Deutschen Herzzentrum der Charité (DHZC).