Hypoplastisches Linksherzsyndrom (HLHS)
Beim Hypoplastischen Linksherzsyndrom (HLHS) sind die Strukturen des linken Herzens zu klein ausgebildet. Sie sind so klein, dass die linke Herzkammer nicht oder nur teilweise zur Versorgung des Körperkreislaufes beitragen kann.
Ursache
Die linke Herzkammer konnte sich nicht richtig entwickeln. Ursache dafür ist eine Unterentwicklung (Hypoplasie) oder ein vollständiges Fehlen (Aplasie) der Kammer.
Dahinter steckt meist eine schwere Einengung oder ein völliger Verschluss der Mitralklappe (Mitralstenose bzw. -atresie). Über diese Klappe fließt das Blut normalerweise in die linke Herzkammer.
Auch eine Verengung oder ein Verschluss der Aortenklappe (Aortenstenose bzw. -atresie) kann die Entwicklung der linken Kammer verhindern. Diese Klappe ist dafür zuständig, dass das Blut aus der linken Kammer in den Körperkreislauf gepumpt wird.
Weiterhin besteht oft eine Unterentwicklung der aufsteigenden Schlagader (Aorta) und des Aortenbogens, die Kammerscheidewand ist aber in den meisten Fällen intakt.
Heute wird der Begriff HLHS auch für andere Formen der zu kleinen linken Herzkammer angewandt, nämlich dann, wenn die linke Kammer nicht den Körperkreislauf unterstützen kann, sondern die rechte Kammer diese Aufgabe übernehmen muss. Die rechte Herzkammer wirft das Blut nun über die Lungenschlagader in die Lunge, aber auch über eine Kurzschlussverbindung (den sogenannten Ductus arteriosus) in den Körperkreislauf aus. Das Problem besteht darin, dass der Ductus sich normalerweise nach der Geburt verschließt und dass beim HLHS damit der Körper praktisch nicht mehr durchblutet wird. Daher muss das Neugeborene sofort nach der Geburt mit Medikamenten behandelt werden, die den Ductus offenhalten. Das ist meist auch gut möglich, da die Diagnose pränatal im Rahmen einer Feindiagnostik schon im 2. Drittel der Schwangerschaft gestellt werden kann.
Bereits vor der Geburt bieten wir nach Diagnosestellung auch eine ausführliche Beratung der werdenden Eltern über die therapeutischen Optionen an. Kontaktieren Sie unser Sekretariat zur Terminvereinbarung gern telefonisch oder per E-Mail:
Daniela Peters, Sylvia Evers und Mandy Müller
+49 30 2493 3400
kinderherzchirurgie@dhzc-charite.de

Bereits vor der Geburt bieten wir nach Diagnosestellung auch eine ausführliche Beratung der werdenden Eltern über die therapeutischen Optionen an. Kontaktieren Sie unser Sekretariat zur Terminvereinbarung gern telefonisch oder per E-Mail:
Daniela Peters, Sylvia Evers und Mandy Müller
+49 30 2493 3400
kinderherzchirurgie@dhzc-charite.de
Nach der Entbindung sollte das Kind idealerweise unmittelbar durch eine:n Neonatolog:in versorgt und die Diagnose HLHS durch eine:n Kinderkardiolog:in echokardiographisch gesichert werden. Nach Versorgung des Neugeborenen sollte innerhalb der ersten Lebenstage die Verlegung in ein ausgewiesenes Zentrum zur operativen Versorgung erfolgen (die Kinderherz-Notfall-Hotline des DHZC erreichen Sie telefonisch unter +49 30 4593 2836; kinderkardiologische Intensivstation).
Symptome
Kinder mit einem Hypoplastischen Linksherzsyndrom (HLHS) sind vor der Operation meist zyanotisch (blau) oder sehr blass, was ein Zeichen für eine schlechte periphere Durchblutung ist. Außerdem sieht man häufig eine schnelle und angestrengte Atmung, einen sehr schnellen Herzschlag sowie schwach tastbare Pulse.
Diagnose
Die Diagnose wird während der Schwangerschaft im Pränatalscreening (Feinultraschalluntersuchung) oder nach der Geburt mittels Echokardiographie durch eine:n Kinderkardiolog:in gestellt. Ergänzend kann auch eine Herzkatheteruntersuchung bei unklaren Befunden erforderlich sein.
Therapie
Norwood-Operation
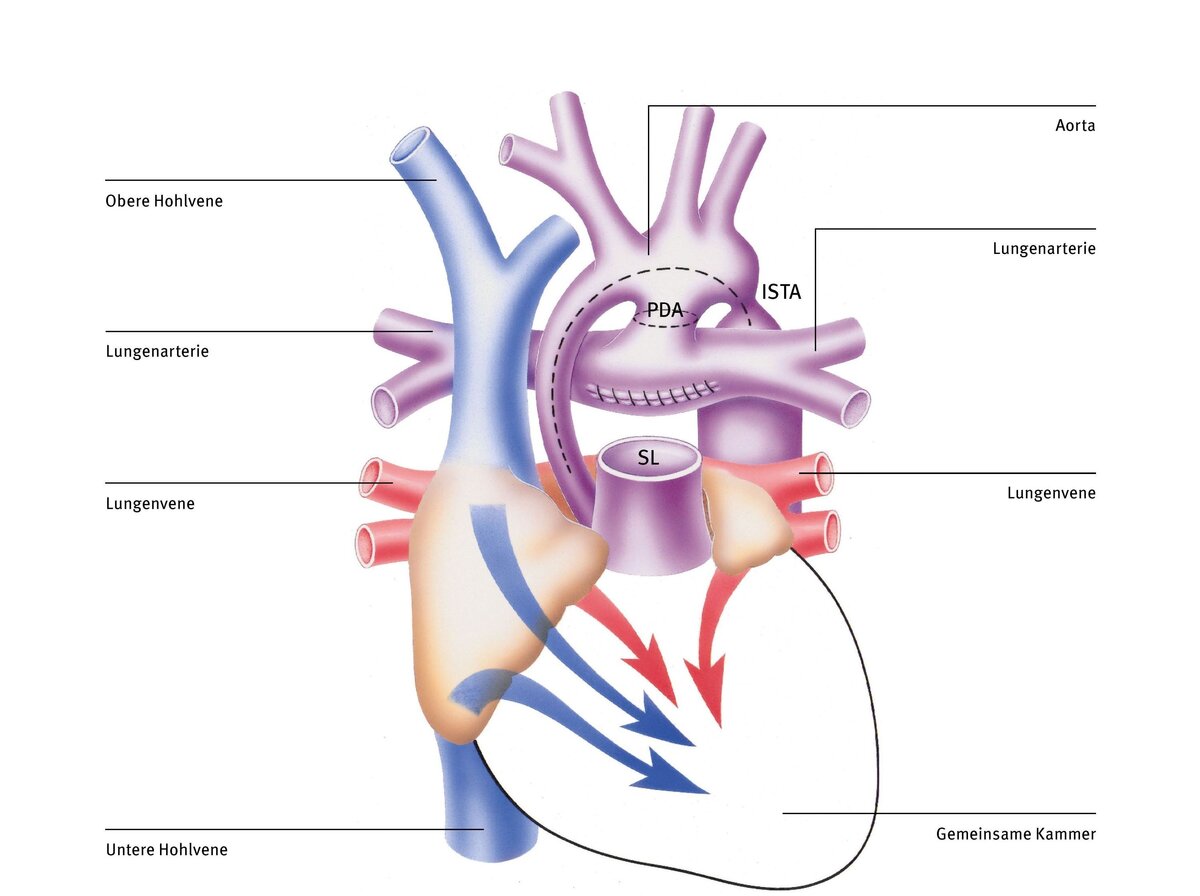
Die Norwood-Operation wird schon in den ersten Lebenstagen als lebensrettende Erstmaßnahme durchgeführt. Sie ist die erste von drei Operationen, die darauf abzielen, den Blutkreislauf so umzustrukturieren, dass die rechte Herzkammer als einzige Pumpkammer (da ja die linke Kammer fehlt) das ganze Blut sowohl in die Lunge als auch in den restlichen Körper pumpt – eine schwere Aufgabe.
Dabei wird nach Anschluss an die Herz-Lungen-Maschine zunächst der Stamm der Lungenschlagader (Pulmonalarterie, PA) vom Herzen abgetrennt. Anschließend wird die Kurzschlussverbindung von der Lungenarterie zur Aorta (persistierenden Ductus arteriosus, PDA) durchtrennt (gestrichelte Linie). Nun werden die viel zu kleine aufsteigende Körperschlagader (Aorta) und der gesamte Aortenbogen bis über die Engstelle (Aortenisthmusstenose, ISTA) aufgeschnitten (gestrichelte Linie).
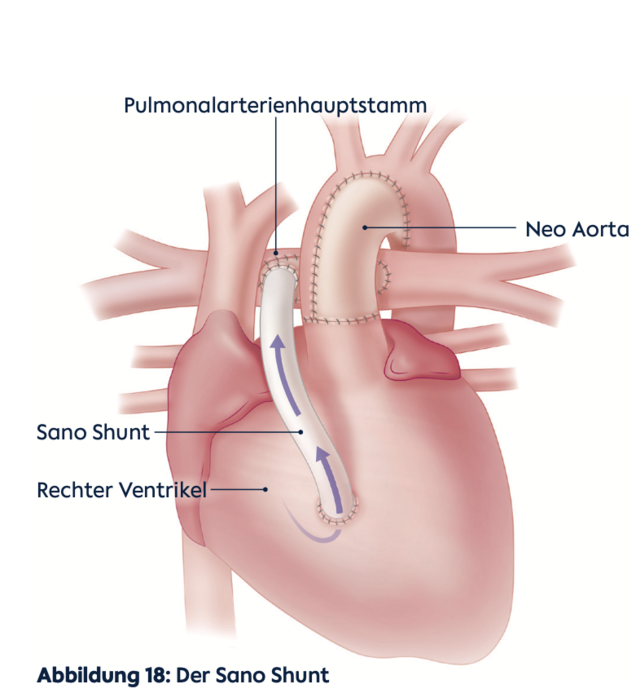
Um zu gewährleisten, dass der Körperkreislauf ausreichend mit Blut versorgt wird, vergrößert man den aufgeschnittenen Teil der Aorta mit einem Flicken und verbindet ihn mit dem Stamm der Lungenarterie und der Wurzel der Aorta, die man zuvor zusammengenäht hat (Dames Kaye Stansel Anastomose, DKS). Dadurch werden der Anfangsteil der Aorta und die Engstelle hinter dem Aortenbogen (ISTA) ausreichend erweitert. Die rechte Pumpkammer kann jetzt das Blut ohne Widerstand in den Körper auswerfen. Zusätzlich wird ein kleines Kunststoffröhrchen (Shunt) zwischen einem größeren Ast der Aorta und der Lungenarterie eingenäht, um ausreichend, aber nicht zu viel Blut in die Lunge zu leiten. Alternativ kann die Lunge auch durch ein Röhrchen versorgt werden, das direkt mit der rechten Herzkammer verbunden wird (Sano Shunt Variante).
Das Mischblut fließt nach der Norwood-Operation ungehindert in die rechte Herzkammer und von dort auch ungehindert in die Aorta, da jetzt der zu kleine Anfangsteil und die Verengung hinter dem Aortenbogen mit einem Flicken erweitert wurde. Die Blutversorgung der Lunge ist mittels des Kunststoffröhrchens zwischen der Aorta (oder der rechten Kammer) und der Lungenarterie sichergestellt.
Nach Erholung von der Norwood-Operation folgen weitere Operationen im Alter von vier bis sechs Monaten (Glenn-Operation) sowie im Alter von zwei bis drei Jahren (Fontan-Operation), mit denen der Lungen- und Körperkreislauf schrittweise getrennt wird und damit die Belastung für die rechte Herzkammer erheblich abnimmt.
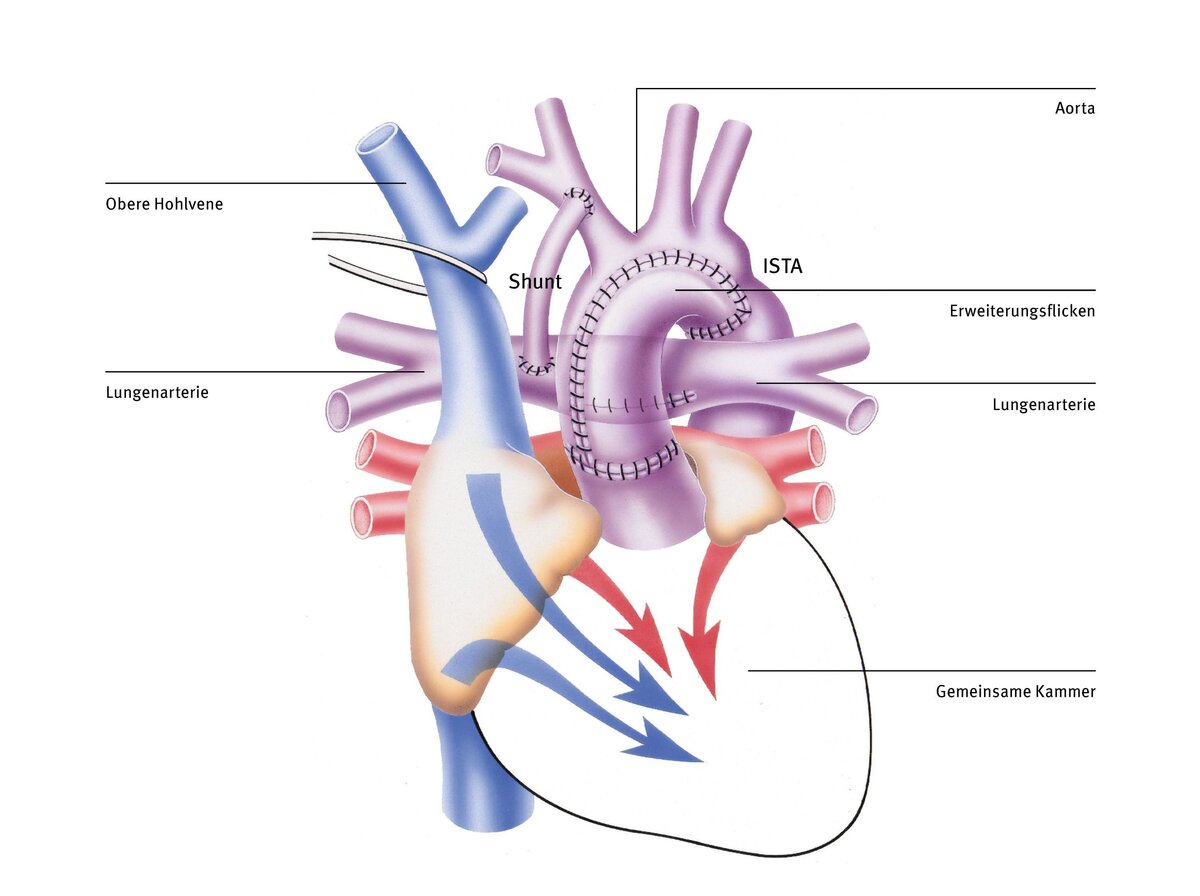
Bidirektionale Glenn-Operation (obere cavopulmonale Anastomose)
Die rechte Herzkammer, die normalerweise nur Blut in den Lungenkreislauf auswirft, versorgt nach der Norwood-Operation den gesamten Körper mit Blut. Für längere Zeit wäre diese Herzkammer damit sicher überfordert, denn normalerweise versorgt sie ja nur den Lungenkreislauf und nicht den Körperkreislauf (diese Aufgabe wird im normalen Herzen von der linken Herzkammer erledigt). Ein weiterer Nachteil ist, dass in beiden Kreisläufen nur gemischtes Blut fließt.
Mit der bidirektionalen Glenn-Operation (auch obere cavopulmonale Anastomose genannt) wird der Körper- und Lungenkreislauf teilweise getrennt. Dabei wird das Kunststoffröhrchen entfernt und die obere Hohlvene auf Höhe des rechten Vorhofs abgetrennt und direkt mit der rechten Lungenschlagader verbunden. Durch die Operation wird die rechte Herzkammer, die bisher eine erhöhte Arbeitsbelastung hatte, teilweise entlastet. Diese Anastomose kann aber erst nach Abfall des Gefäßwiderstandes in den Lungengefäßen im Alter von drei bis sechs Monaten durchgeführt werden. Dann kann das Blut passiv durch die Lunge fließen, nur angetrieben von der Schwerkraft und durch den Sog, der beim Einatmen entsteht.
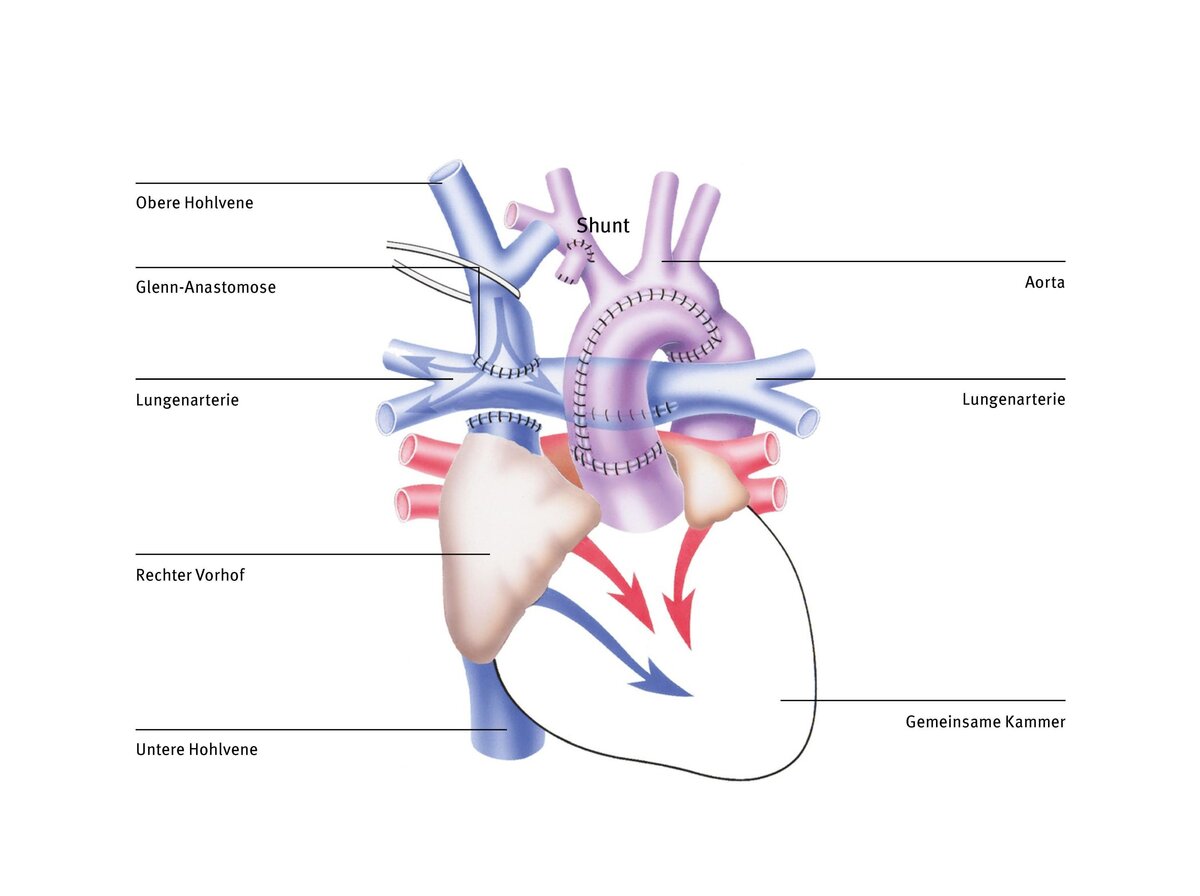
Fontan-Operation (totale cavopulmonale Anastomose)
Bei der Glenn-Operation wird erreicht, dass das sauerstoffarme Blut aus der oberen Hohlvene direkt in die Lunge fließt und sich nicht mehr mit dem sauerstoffreichen Blut aus der Lunge im gemeinsamen Herzvorhof mischt. Das Blut aus der unteren Hohlvene ist jedoch im Säuglingsalter noch nicht umgeleitet worden, weil die Lungengefäße noch nicht „groß genug“ waren bzw. der Gefäßwiderstandes in den Lungengefäßen noch zu hoch war, um das gesamte Blut aus dem Körperkreislauf aufzunehmen. Um die einzige Herzkammer besser zu entlasten und damit auch Lungen- und Körperkreislauf komplett zu trennen, muss das Blut aus der unteren Hohlvene ebenfalls direkt in die Lunge, statt in die gemeinsame Herzkammer geleitet werden.
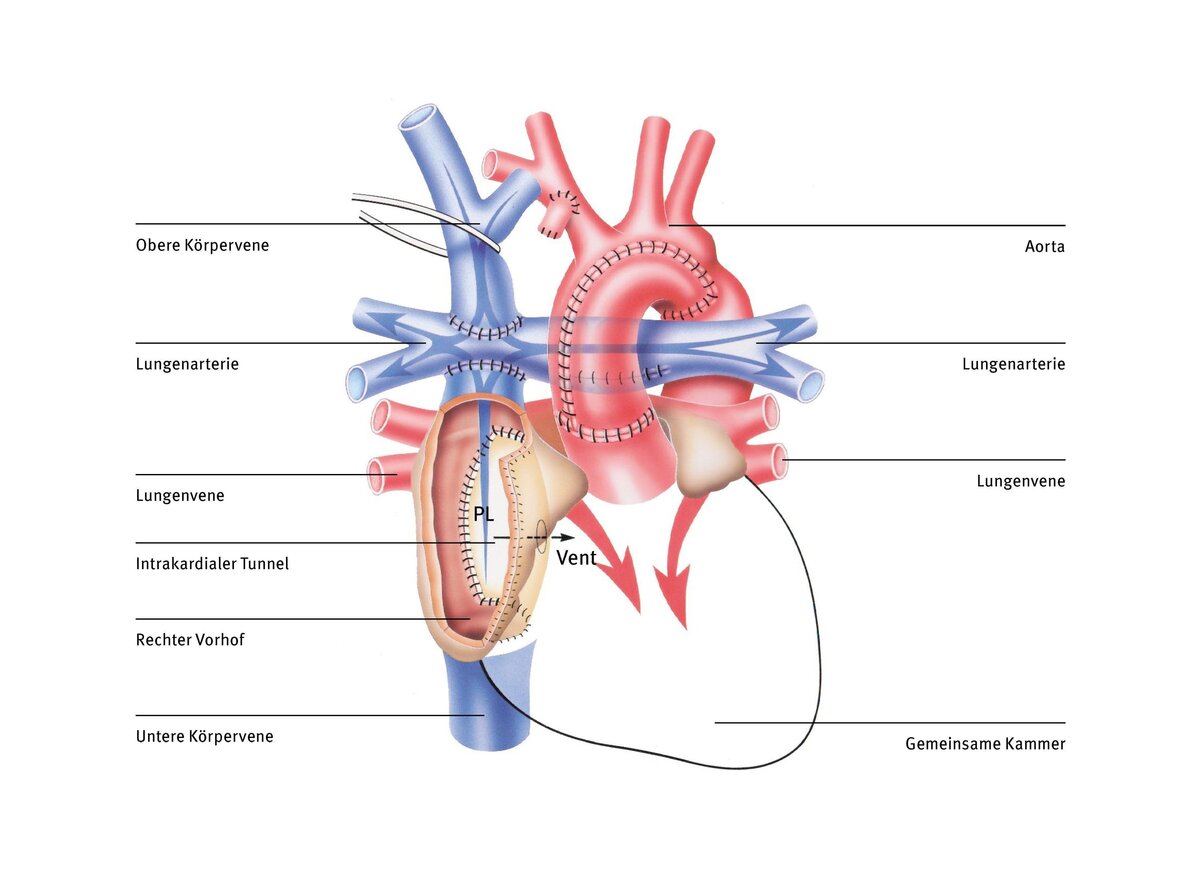
Hierbei gibt es verschiedene Techniken. Die heute am häufigsten verwendete ist die totale cavopulmonale Anastomose (TCPC), bei der eine Gefäßumleitung (Tunnel) außen und rechts am Herzen vorbeigeführt und so das Blut der unteren Hohlvene direkt der Lunge zugeführt wird. Diese Art der Zuleitung des Venenblutes zur Lunge wurde zuerst von dem französischen Herzchirurgen Francis Fontan in den 1980er Jahren entwickelt. Heute nennt man den so geschaffenen Umgehungskreislauf, bei der das gesamte Körpervenenblut passiv durch die Lunge fließt, auch Fontankreislauf.
Jetzt sind Lungen- und Körperkreislauf ganz voneinander getrennt und es fließt kein Mischblut mehr in der Aorta. Sauerstoffarmes Blut fließt über die obere und untere Hohlvene direkt in die Lungenarterie. Das sauerstoffreiche Blut aus der Lunge wird von der Herzkammer in die Aorta gepumpt. Die Herzkammer muss nach der Fontan-Operation nur noch das Blut aus der Lunge in den Körperkreislauf pumpen.
Therapie am Deutschen Herzzentrum der Charité
Über die letzten zehn Jahre hat sich das Deutsche Herzzentrum der Charité zu einem führenden Zentrum für die Behandlung des Hypoplastischen Linksherzsyndroms entwickelt. Die Überlebensrate der Norwood-Operation liegt derzeit bei über 90 %, ein im internationalen Vergleich herausragendes Ergebnis. Verschiedene Behandlungs- und Operationstechniken wurden laufend angepasst und verbessert.
Bei hämodynamischer Stabilität und Infektionsfreiheit führen wir die Norwood-Operation schon in den ersten vier Lebenstagen durch. Das frühe OP-Timing soll das mögliche Entstehen von präoperativen Komplikationen verhindern und ist mit einem besseren postoperativen Outcome nach Norwood-Palliation verbunden (Anderson BR et al., 2015).
Wir führen heute die Operation nicht mehr im Kreislaufstillstand mit Abkühlen der Patient:innen auf unter 18°C durch, sondern mit einer Temperaturabsenkung nur bis 28°C und kontinuierlicher Durchblutung der oberen und unteren Körperhälfte über spezielle Kanülen in den Schlagadern der Kopf- und Beingefäße. Die Methode ist relativ einfach, schnell und komplikationsarm und erlaubt die durchgängige Durchblutung des Gehirns und der Bauchorgane während der Phase der Aortenbogenrekonstruktion (Boburg et al.).
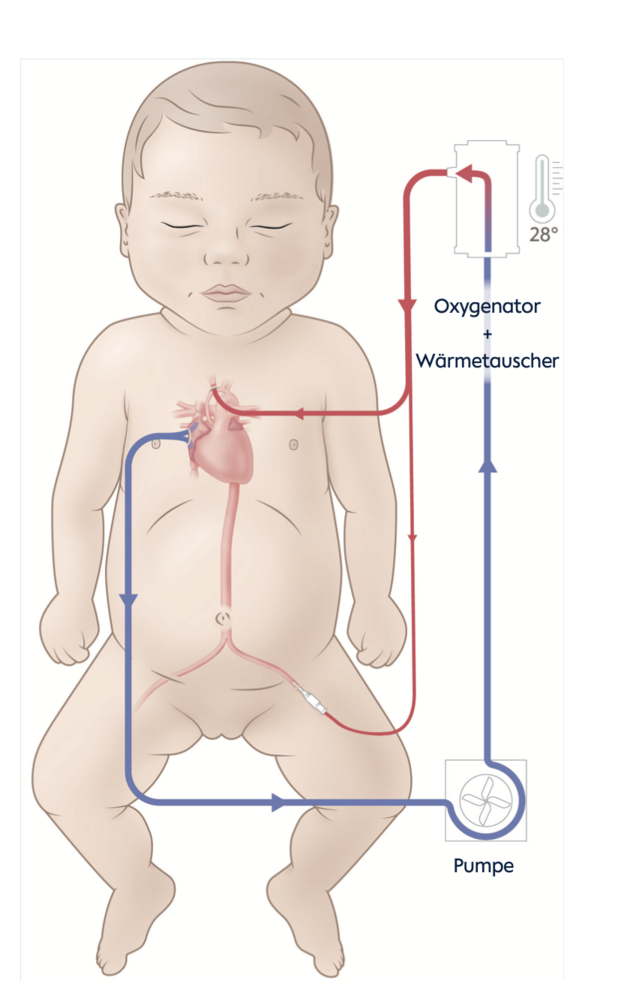
Des Weiteren haben wir die Shunt-Präferenzen gewechselt: von der modifzierten Blalock Thomas Taussig Variante (BTT, mit einem Kunststoffshunt von der rechten Kopf-und Armschlagader auf die rechte Lungenschlagader) zum jetzt häufiger verwendeten Sano-Shunt (von der rechten Herzkammer zur Lungenschlagader). In einer großen Vergleichsstudie hat sich neben der schon bekannten stabileren Kreislauflage auch ein Überlebensvorteil bis zum Alter von fünf Jahren für diese Variante gezeigt (Newburger et al. 2014). Danach gleichen sich die Überlebenskurven von BTT und Sano-Shunt an.
Das reine Überleben ist allerdings nur ein Behandlungsergebnis, eine verbesserte körperliche und geistige Entwicklung ein weiteres bedeutendes. Da wir eine wesentlich stabilere Kreislauflage und einen kürzeren und komplikationsärmeren postoperativen Verlauf in den ersten vier bis sechs Monaten bis zur Glenn-Operation mit der Sano Shunt Variante sehen, ist auch mit einer besseren Entwicklung zu rechnen. Dies wird durch eine Studie zur intellektuellen Entwicklung in Beziehung mit der Kreislaufstabilität der Säuglinge belegt (Newburger et al. 2012).
Die BTT Shunt-Variante kommt so derzeit nur noch bei besonderen Begebenheiten, z.B. Trikuspidalklappenatresie mit Transpositionsstellung und hypoplastischem Aortenbogen, zur Anwendung, weil sich kaum Platz für das Annähen des Sano-Shunts an der zu kleinen Auslasskammer findet bzw. nicht mit einer zuverlässigen Blutversorgung aus der Auslasskammer zu rechnen ist.
Das Homemonitoring mit EVIE App senkt die Sterberate zwischen der ersten Norwood-Operation und der nachfolgenden Glenn-Operation (Interstage) auf nahezu null. Alle Kinder erhalten einen Monitor, womit die Eltern Sauerstoffsättigung, das Körpergewicht, die Herzfrequenz und die Temperatur nach Messung eintragen und mit der Smartphone-App Evie an das DHZC übermitteln können. Die Daten werden täglich und sofort übermittelt und die Eltern bei Auffälligkeiten umgehend kontaktiert. Falls nötig, werden die Säuglinge zeitnah stationär aufgenommen.

Video: Eine Mutter erzählt
Salih kam mit einem hypoplastischen Linksherz-Syndrom zur Welt und wurde am DHZC erfolgreich behandelt. In unserem Video spricht seine Mutter Gülcan I. offen darüber, wie sie die Zeit von der Diagnose bis zur dritten Operation erlebt hat.
Prognose
Die Prognose des Kindes ist bei dem zugrundeliegenden Herzfehler und mit einem singulären Ventrikel kurz- bis mittelfristig gut. Meist können sich die Kinder gut und altersentsprechend belasten.
Die neuromotorische Entwicklung hängt von vielen Faktoren ab, darunter etwa Frühgeburtlichkeit und Kreislaufinstabilitäten.
Die langfristige Prognose ist schwer abschätzbar und individuell unterschiedlich, sie hängt von Entwicklung und Ausmaß der bekannten Langzeitkomplikationen (Eiweißverlustenteropathie, Herzrhythmusstörungen, Herzversagen) ab.
Quellenangaben
- Anderson BR et al. Earlier stage 1 palliation is associated with better clinical outcomes and lower costs for neonates with hypoplastic left heart syndrome. J Thorac Cardiovasc Surg. 2015;149(1):205-10.e1.
- Boburg et al. Selective lower body perfusion during aortic arch surgery in neonates and small children. Perfusion 2020;35:621–5.
- Newburger et al. Circulation. 2014;129:2013-2020 Newburger et al. Circulation. 2012 May 1; 125(17): 2081–2091.
- Rosenthal et al. Application-based remote interstage home monitoring for infants with shunt- or duct-dependent pulmonary perfusion. Front Cardiovasc Med 2025;11:1493698.
Autor

Prof. Dr. med. Joachim Photiadis ist Direktor der Klinik für Chirurgie Angeborener Herzfehler – Kinderherzchirurgie am Deutschen Herzzentrum der Charité (DHZC) und Mitglied des Erweiterten Bereichsvorstands des DHZC.