ALCAPA-Syndrom / ARCAPA-Syndrom
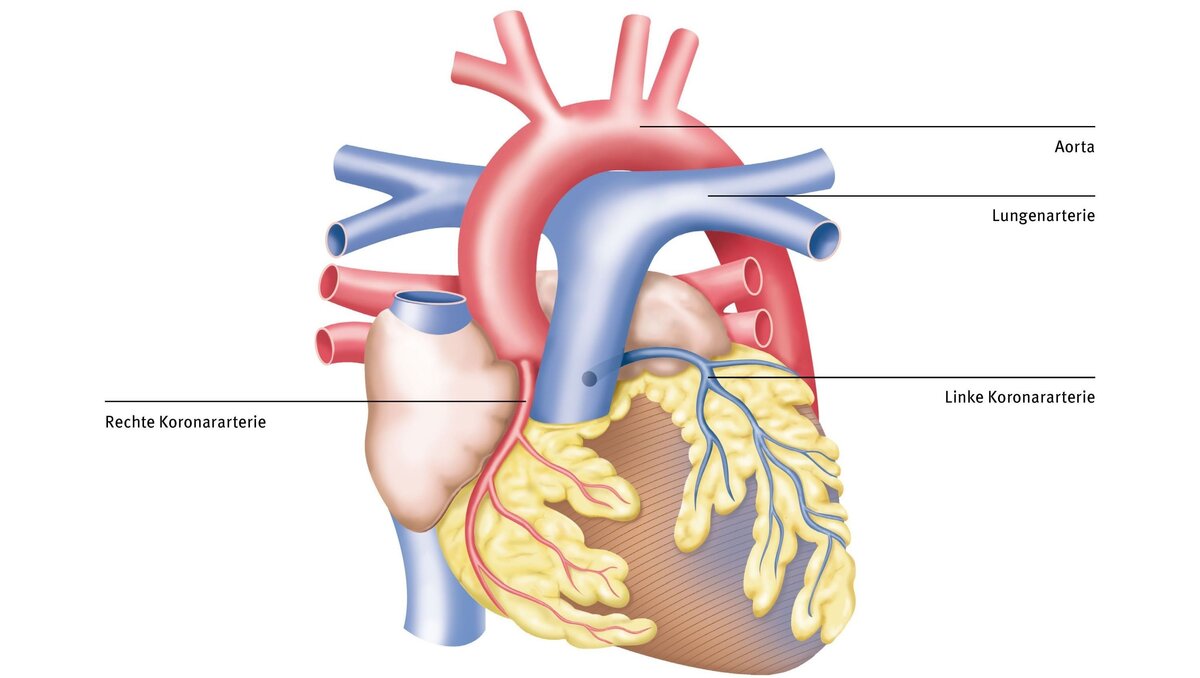
Das ALCAPA-Syndrom (Anomalous Left Coronary Artery from the Pulmonary Artery), welches auch als Bland-White-Garland-Syndrom bezeichnet wird, ist ein sehr seltener angeborener Herzfehler – er betrifft etwa 0,5 % aller angeborenen Herzfehler. Dabei entspringt die linke Herzkranzarterie von der Lungenschlagader anstatt der Hauptschlagader (Aorta). Beim ARCAPA-Syndrom (Anomalous Right Coronary Artery from the Pulmonary Artery) entspringt die rechte Herzkranzarterie von der Lungenschlagader anstatt von der Aorta.
Dies hat zwei Folgen: In den ersten Lebenswochen sinkt bei allen Babys der Druck in der Lungenschlagader ab. Der Druck in der Lungenschlagader ist dann viel niedriger als in der Hauptschlagader, aus der normalerweise die Herzkranzgefäße entspringen.
Das führt bei Kindern mit einem ALCAPA-Syndrom häufig dazu, dass im Alter von wenigen Wochen bis Monaten das linke Herzkranzgefäß den Herzmuskel nicht mehr ausreichend mit Blut versorgt.
Ursachen und Auswirkungen
Diese Unterversorgung mit Blut, Sauerstoff und Nährstoffen kann zu einer Herzschwäche und einer erheblichen Herzfunktionseinschränkung sowie bei längerem Bestehen dieser Sauerstoffarmut auch zu einem irreversiblen Untergang von Herzmuskel (Herzinfarkt) führen.
Die linke Herzkranzarterie versorgt vorwiegend die linke Herzkammer, die das sauerstoffreiche Blut über die Hauptschlagader in den Körper pumpt. Eine Herzschwäche in diesem Bereich ist daher besonders kritisch, weil diese ein Kreislaufversagen nach sich ziehen kann. Abhängig von den ausgebildeten Kollateralen (also den Gefäßverbindungen von der normal aus der Aorta entspringenden rechten Herzkranzarterie zur falsch entspringenden linken Herzkranzarterie) variieren die Symptome der Patient:innen. Bei schlecht ausgebildeten Kollateralen kann es schon im Alter von wenigen Lebenswochen, wenn der Lungengefäßwiderstand sinkt, zu schwerwiegenden Symptomen kommen. Bei anderen Patient:innen mit guter Kollateralversorgung kann ein ALCAPA-Syndrom hingegen auch lange unbemerkt bleiben.
Risikofaktoren
Das ALCAPA-Syndrom entsteht durch die fehlerhafte Anlage / Ursprung des linken Herzkranzgefäßes wahrscheinlich schon in den ersten drei Schwangerschaftsmonaten des ungeborenen Kindes. Die Gründe für diese Fehlentwicklung sind nicht geklärt. Man nimmt an, dass viele verschiedene Faktoren und Umwelteinflüsse in der Schwangerschaft eine Rolle spielen können.
Meistens tritt das ALCAPA-Syndrom als isolierter Herzfehler auf, was bedeutet, dass es selten begleitende Fehlbildungen im Herzen gibt. Allerdings besteht aufgrund der eingeschränkten Funktion der linken Herzkammer häufig auch eine unterschiedlich ausgeprägte Undichtigkeit der Mitralklappe.
Symptome
Das ALCAPA-Syndrom führt meist in den ersten Lebenswochen bis -monaten zu einer Unterversorgung der linken Herzkammer, die dadurch das Blut nicht mehr ausreichend aus der Lunge in den Körper pumpen kann. Die Kinder zeigen Symptome (Krankheitszeichen) einer Herzschwäche mit schneller Atmung, Schwitzen, schnellem Ermüden und Trinkschwäche.
Eine eingeschränkte Pumpfunktion und Erweiterung der linken Herzkammer führt häufig dazu, dass die Mitralklappe nicht mehr richtig schließt und so ein guter Teil des Blutes der linken Herzkammer nicht in den Körperkreislauf, sondern zurück in den linken Vorhof ausgeworfen wird. Das Blut staut sich in die Lunge zurück, was die Symptome noch verstärken kann. Bei einer starken Einschränkung der Pumpfunktion kann das auch schnell zu einer lebensbedrohlichen Situation führen, wenn das Herz kaum noch Blut in den Körper zur Versorgung der Organe pumpen kann („Low Cardiac Output“). Dies äußert sich durch Atemnot und erhebliche Belastungseinschränkung. Eine Ausdünnung der Herzwand (Aneurysma) durch das Zugrundegehen von Herzmuskulatur ist ebenfalls möglich.
Durch die Sauerstoffunterversorgung des Herzmuskels oder des Reizleitungssystems ist das Risiko für Herzrhythmusstörungen erhöht, die unter Umständen zu einem plötzlichen Herztod führen können. Der Herzfehler sollte daher in jedem Lebensalter auch bei geringen Symptomen zeitnah behoben werden.
In Ausnahmefällen kann sich der Herzfehler bei sehr guter Kollateralversorgung auch erst später, teilweise sogar erst im Erwachsenenalter, bemerkbar machen. In diesen Fällen können sich die Symptome wie z.B. `Druck auf der Brust´ erst später äußern.
Oft kann schon mittels der Echokardiographie die Verdachtsdiagnose gestellt werden. Mit einer Herzkatheteruntersuchung lässt sich das ALCAPA-Syndrom zuverlässig bestätigen. Sie zeigt den Ursprung der linken Koronararterie aus der Pulmonalarterie anstelle der Aorta.
Oft kann schon mittels der Echokardiographie die Verdachtsdiagnose gestellt werden. Mit einer Herzkatheteruntersuchung lässt sich das ALCAPA-Syndrom zuverlässig bestätigen. Sie zeigt den Ursprung der linken Koronararterie aus der Pulmonalarterie anstelle der Aorta.
Diagnose
Das ALCAPA-Syndrom ist eine seltene angeborene Fehlbildung der Koronararterien. Die Diagnose erfolgt durch eine Kombination aus klinischer Untersuchung, Bildgebung und gegebenenfalls weiteren Tests.
Diagnostische Methoden:
- Die Echokardiographie (Herzultraschall) ist das erste bildgebende Verfahren zur Diagnose. Sie zeigt eine erweiterte linke Herzkammer und eine schlechte Kontraktionsleistung (Anzeichen einer Ischämie).
- Die Herzkatheteruntersuchung (Koronarangiographie) ist der Goldstandard zur Bestätigung der Diagnose. Sie zeigt den Ursprung der linken Koronararterie aus der Pulmonalarterie anstelle der Aorta.
- Das EKG (Elektrokardiogramm) kann Anzeichen einer Myokardischämie oder eines früheren Herzinfarkts zeigen.
- Eine Pulsoxymetrie und Blutgasanalyse können bei Neugeborenen eine verminderte Sauerstoffsättigung zeigen, wenn eine schwere Ischämie vorliegt.
- Das Herz-MRT oder die CT-Angiographie ermöglicht eine detaillierte Darstellung der Koronargefäße und deren Anomalien. Damit lässt sich die Myokarddurchblutung beurteilen. Meist ist eine MRT- und CT-Untersuchung jedoch entbehrlich, da nach Diagnosestellung zeitnah eine Korrekturoperation geplant werden sollte.
Therapie und Operation
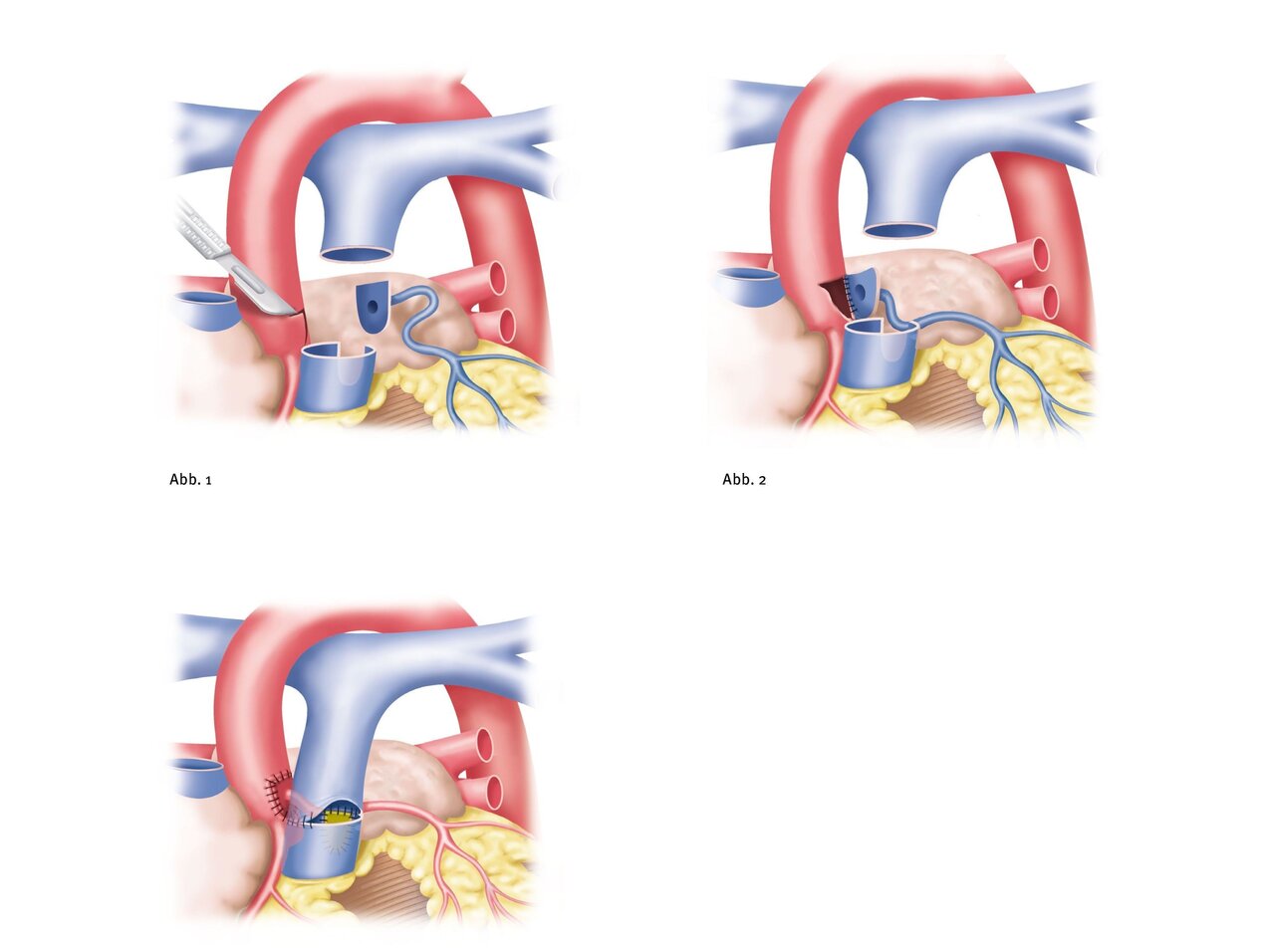
Ein ALCAPA-Syndrom wird normalerweise durch einen sogenannten „Herzkranzgefäßtransfer“ behandelt. Dabei wird die falsch entspringende linke Herzkranzarterie von der Lungenarterie in die Hauptschlagader eingesetzt. Der Eingriff wird nach Eröffnen des Brustbeins unter Verwendung der Herz-Lungen-Maschine und im Herzstillstand durchgeführt. Dabei wird die fehlabgehende linke Herzkranzarterie an ihrem Ursprung aus der Lungenschlagader mit etwas Randsaum zum Nähen ausgeschnitten. Anschließend wird das Gefäß vorsichtig etwas aus dem Gewebe gelöst, um es beweglicher zu machen.
Es ist wichtig, dass kein Zug auf das umgesetzte Herzkranzgefäß kommt, da es ansonsten erneut zur Engstellung des Herzkranzgefäßes und damit zur Sauerstoffunterversorgung von Teilen des Herzens kommen kann. Danach wird die Herzkranzarterie an einer passenden Stelle in der Hauptschlagader wieder eingenäht. Das durch das Ausschneiden der Herzkranzarterie entstandene „Loch“ in der Lungenschlagader wird mit einem kleinen Flicken aus eigenem Herzbeutel wieder verschlossen.
Meist besteht zum Zeitpunkt der Operation bereits eine leicht- bis mittelgradige Undichtigkeit der Mitralklappe. Eine geringe Undichtigkeit verbessert sich normalerweise erst Wochen nach der Operation durch die verbesserte Sauerstoffzufuhr und dadurch bessere Funktion des Herzmuskels. Ist die Undichtigkeit allerdings hochgradig, kann es nötig werden, dass in derselben Operation eine Reparatur der Mitralklappe vorgenommen werden muss.
Liegt bereits vor der Operation durch die chronische Sauerstoffminderversorgung eine deutlich eingeschränkte Pumpfunktion der linken Kammer vor (LVEF < 35%), so kann diese auch nach erfolgreicher Operation zunächst bestehen bleiben. In einigen Fällen bedarf es des Einsatzes eines mechanischen Kreislaufunterstützungssystems, um einen ausreichenden Kreislauf nach dem Eingriff herzustellen. Das Herzunterstützungssystem kann jedoch in den meisten Fällen nach wenigen Wochen Erholungszeit des Herzens und mit wieder hergestellter Pumpfunktion entfernt werden.
Therapie am DHZC
Da es sich beim ALCAPA-Syndrom um einen sehr seltenen angeborenen Herzfehler handelt und für die erfolgreiche Behandlung das Umsetzen der Herzkranzarterie technisch einwandfrei erfolgen muss, sollte eine Behandlung der Patient:innen in Zentren mit großer Expertise erfolgen.
Am Deutschen Herzzentrum der Charité (DHZC) haben wir große Erfahrung mit der Versorgung von Kindern und Erwachsenen mit ALCAPA-Syndrom und auch zu diesem Herzfehler bereits viele wissenschaftliche Artikel veröffentlicht. Da wir auch bei anderen Herzfehlern die Herzkranzgefäße schon im Neugeborenenalter und auch bei schwierigen Verhältnissen (Koronaranomalien) umsetzen, führen wir den „Koronartransfer“ routiniert und mit sehr gutem Ergebnis durch.
Sollte im seltenen Fall die Herzschwäche auch nach Korrekturoperation weiterbestehen, stehen bei uns Herzunterstützungssysteme in jeder Größe zur Verfügung. Das Kunstherz-Behandlungsteam am DHZC zeichnet sich durch die weltweit größte Erfahrung beim Einsatz, im Umgang und der Entfernung mit solchen Kunstherzsystemen bei Kindern und Erwachsenen aus. Die große Routine bei einem so seltenen und komplexen Herzfehler macht sich im sehr guten postoperativen Verlauf unserer Patient:innen bemerkbar. Weitere Informationen dazu gibt es in unseren Qualitätsstandards, unserer extern validierten Qualitätssicherung und in unseren Qualitätsjahresberichten.
Nachsorge
Konnte eine rechtzeitige Korrektur mit Umsetzen der linken Koronararterie auf die Hauptschlagader ohne erhebliche Funktionseinschränkungen der linken Herzkammer erfolgen, so ist nach dem Krankenhausaufenthalt und einer Erholungszeit von zirka einem halben Jahr grundsätzlich eine normale Belastung und damit auch eine normale Entwicklung möglich. In den meisten Fällen wird eine zum Zeitpunkt der Operation bestehende geringe bis mäßige Mitralklappenundichtigkeit gut toleriert und ist meistens über die Zeit rückläufig. Dies braucht allerdings Wochen bis Monate, sodass es nach der Operation noch Zeichen der Herzschwäche geben kann, die einer medikamentösen Unterstützung bedürfen. Das Brustbein, welches wir bei dieser Operation längs durchtrennen müssen, braucht in der Regel etwa vier bis sechs Wochen, um wieder vollständig zu verheilen und stabil zu sein.
Zur Kinderherzchirurgie am DHZC
Diagnostische Herzkatheteruntersuchungen am DHZC
In zwei speziell ausgerüsteten und hochmodernen Herzkatheterlaboren der Klinik für Angeborene Herzfehler – Kinderkardiologie untersuchen und behandeln wir Neugeborene, Säuglinge, Kinder und Jugendliche jeden Alters mit angeborenen und erworbenen Herzerkrankungen, sowie auch alle Erwachsenen mit angeborenen Herzfehlern. Wir sind als überregionales Schwerpunktzentrum auf die invasive kardiologische Diagnostik und interventionelle Therapie von Patient:innen mit angeborenen Herzfehlern spezialisiert. Interventionelle Behandlungen im Herzkatheterlabor ergänzen oder ersetzen zunehmend chirurgische Eingriffe und sind entscheidend, um die optimale Therapie der Patient:innen festzulegen. Unser Team verfügt über langjährige Erfahrung und Expertise.
Fragen und Antworten für Eltern (FAQ)
Falls die Undichtigkeit der Mitralklappe weiter zunimmt und es zum Rückstau des Blutes in die Lunge kommt, sind im weiteren Verlauf Eingriffe an der Mitralklappe erforderlich. Diese kann in den meisten Fällen durch eine Reparatur der Klappe erfolgen. Ist eine Reparatur nicht möglich, muss die Klappe ersetzt werden.
Ein zu langsamer Herzrhythmus, der durch eine Minderversorgung von Teilen des Reizleitungssystems zustande kommt, muss eventuell durch eine Schrittmacher-Implantation behoben werden.
Die linksseitige AV-Klappe (Mitralklappe), deren Halteapparat durch die chronische Sauerstoffunterversorgung in Mitleidenschaft gezogen wurde, neigt zu späteren Undichtigkeiten, die durch regelmäßige Ultraschallkontrollen beim niedergelassenen Kinderkardiolog:innen kontrolliert werden sollte. Auch die Funktion der linken Herzkammer sollte nach erfolgter Korrektur weiter im Auge behalten werden.
Konnte eine ALCAPA-Korrektur frühzeitig durchgeführt werden, so stehen einer normalen Lebensqualität und in der Regel auch normalen Lebenserwartung nichts im Wege. Allerdings sind weitere Kontrollen bei niedergelassenen Kinderkardiolog:innen unabdingbar, um diese Lebensqualität aufrechtzuhalten. Eine eventuelle Undichtigkeit an der Mitralklappe sollte früh erkannt werden, um einen Rückstau des Blutes in die Lunge und damit eine erneute Herzinsuffizienz vorzubeugen.
Autor:innen

Prof. Dr. med. Joachim Photiadis ist Direktor der Klinik für Chirurgie Angeborener Herzfehler – Kinderherzchirurgie am Deutschen Herzzentrum der Charité (DHZC) und Mitglied des Erweiterten Bereichsvorstands des DHZC.

Dr. med. Victoria Weixler ist Fachärztin der Klinik für Chirurgie angeborener Herzfehler - Kinderherzchirurgie am Deutschen Herzzentrum der Charité (DHZC).